
In 2011, ophthalmologist Jayakrishna Ambati of the University of Virginia and his colleagues made a curious observation: In the pigmented retinal layers of human eye samples afflicted with an advanced form of age-related macular degeneration (AMD), they discovered high concentrations of Alu transcripts. Alu is a class of transposable elements, DNA bits that jump around the genome through a copy-paste mechanism that occurs in the nucleus. Further studies by the team suggested that the Alu RNA was somehow causing inflammation and cell death, but it was a mystery how.
Earlier this year, Ambati’s team uncovered an important clue. Surprisingly, in cultured human retinal pigmented epithelium cells, they found that DNA copies of Alu can occur in the cell cytoplasm—in the form of complementary DNA (cDNA) made from messenger RNA. In fact, that was the first evidence that human cells are even capable of synthesizing DNA in the cytoplasm. And it was the cytoplasmic Alu cDNA that was to blame for retinal cell death, their results suggested.
See “Human Cells Can Synthesize DNA in Their Cytoplasm”
Now, in a new report published today (September 29) in Science Advances, Ambati and an international team of collaborators confirm that Alu cDNA is present in eye samples from people with geographic atrophy, an advanced form of AMD. They also pin down a novel mechanism through which the cytoplasmic DNA triggers an immune reaction that results in cell death: in essence, it mimics an infection. The findings shine a light on new therapeutic strategies for geographic atrophy, for which no proven treatment exists.
“This is a really cool mechanism, and you need . . . to understand [the] mechanism to design appropriate therapeutics,” remarks Margaret DeAngelis, a molecular biologist and geneticist at the University of Buffalo who wasn’t involved in the study. “However, [as] with anything, you need validation and replication for it to be translational.”
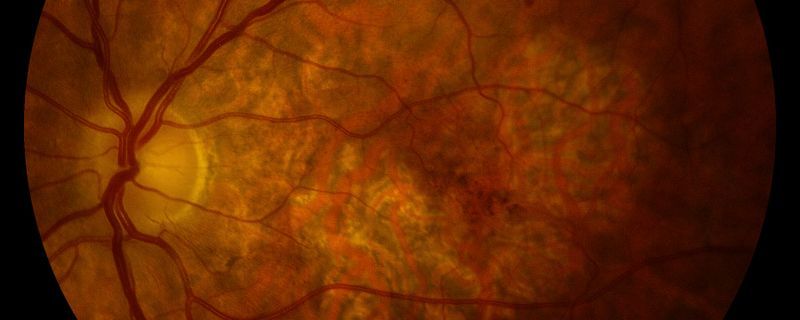
It is a strength of the research, she adds, that it included analysis of real human tissue samples. In the retinal pigmented epithelium of eye samples from 20 deceased people who’d had geographic atrophy, Ambati and his colleagues detected high concentrations of Alu cDNA, which was only faintly detectable in around 20 samples from people without eye disease. Even more striking, Ambati says, was the distribution of the cDNA in the retina. Geographic atrophy is characterized by a gradual, amoeba-like expansion of a lesion that typically begins in the periphery of the retina and eventually consumes the macula, the central patch of the retina that is essential for central vision. The Alu cDNAs “were found exactly at the most active growing edge of the lesions . . . providing a real window of insight into how this is expanding,” says Ambati, who together with several coauthors is listed as an inventor on multiple patents related to the research. He and another coauthor have also received consulting fees from various pharmaceutical companies, and the other coauthor has received research funding from a medical technology company.
His team conducted different experiments to investigate the mechanism whereby Alu cDNA accumulates in such high abundance and could lead to cell death. The molecule’s toxic effect appeared to be dependent on the presence of Alu RNA, and on the heavy activity of LINE 1 (L1)—another group of transposable elements which encodes a reverse transcriptase that converts RNA such as Alu into DNA. The scientists established this, in part, through experiments with the North American rice rat Oryzomys palustris, a species whose genome happens to lack functional L1 copies. The rats were resistant to the degenerative effects of injecting Alu RNA into their eyes, Ambati says. “You put a boatload of this stuff in there, they just laugh at it. Nothing happens to their retina.”
In tests with regular lab mice engineered to lack different DNA sensors, the team found that another component needed for Alu cDNA toxicity is the DNA sensor cyclic guanosine 3′,5′-monophosphate–adenosine 3′,5′-monophosphate (cGAMP) synthase (cGAS), a cellular alarm bell that activates in response to cues such as potentially pathogenic DNA in the cell cytoplasm. Various subsequent experiments—alongside data from the group’s previous work—pointed to a cascade whereby the Alu cDNA’s activation of cGAS disrupts the mitochondrial membrane, triggering mitochondrial DNA to spill into the cytoplasm. The presence of two DNA populations in the cytoplasm activates cGAS again, which in turn triggers the NLRP3 inflammasome, an innate immune system sensor that unleashes a potent inflammatory response, causing cell death.
This relatively straightforward mechanism is “at least to us . . . a very beautiful way in which this is happening, as awful as it is,” Ambati says. As for how the Alu RNA concentration increases to begin with, that could be driven by the age-related decline of a key regulatory enzyme called DICER in the retinal pigmented epithelium, he adds.
Cells of the retinal pigment epithelium don’t regenerate. When they die, the photoreceptor cells they support die too, “so then you lose vision,” says Alfred Lewin, a molecular geneticist at the University of Florida who wasn’t involved in the research. “What’s novel about this [study] is that it’s the first time they found the DNA copies of Alu RNA in eyes donated by patients,” he adds. It’s theoretically conceivable that cells in the human samples were simply senescent, having reached a permanent state of growth cycle arrest in which nuclear DNA is known to leak into the cytoplasm. “But their experiments in [rice] rats show that the presence of this DNA depends on the presence of the RNA and of reverse transcription,” Lewin notes. “Their arguments are pretty sound.”
There are several hypotheses in the AMD field as to what drives the degeneration of retinal tissue, involving oxidative stress, for instance, or lipid deposits that block the flow of nutrients into the retina. Though Lewin says he’s long thought that oxidative stress is likely the driving mechanism, the new data makes a compelling case that Alu cDNA-driven inflammation could play an important role in people with AMD, he says. “I can’t argue with data,” he remarks. “I think it was very detailed, it was very convincing.”
The fact that Alu genetic elements could be underlying the driving mechanism of AMD “is interesting and novel,” DeAngelis says, adding, “I don’t think it’s going to be one disease mechanism.” AMD is a complex disease, with multiple genes, pathways, and environmental factors involved. In addition, the study only examined geographic atrophy, which some consider the late stage of the “dry” form of AMD, characterized by a drying and slimming of the macula. That form is distinct from “wet” AMD, in which blood and other fluids leak into the macula. But there is still no clear consensus among experts about how many stages and types of AMD exist, she notes. “We don’t know if it’s one progressive disease that starts from very early stages . . . or if it’s [multiple] different diseases,” DeAngelis adds.
To Ambati, there is only one good thing about AMD: that it happens over many years and tends to begin in the periphery of vision before it progresses to the center. That gives clinicians a window of around 24 to 30 months to stop the disease’s progression and prevent blindness, says Ambati, who is cofounder of the company Inflammasome Therapeutics, which develops treatments for AMD and other degenerative conditions.
In the new study, he and his colleagues also found that the HIV drug lamivudine—which appeared to block the L1 reverse transcriptase as well as inflammasome signaling—protected mice against the effects of injecting Alu cDNA. And treating mice with Kamuvudines—experimental drugs Ambati and colleagues developed that only inhibit inflammasome activity—had the same effect. Ambati says he thinks that Kamuvudines are a better approach to treating AMD, as they have fewer side effects than HIV treatments.
To that end, he and his colleagues are planning clinical trials in early 2022 to test Kamuvudines in patients in the early phases of geographic atrophy, he says. “It’s well past time that we have some treatments for this condition. God knows we need it.”
[“source=the-scientist”]
